Single-Cell Analysis Copilot
Analyze scRNA-seq data end-to-end — QC, clustering, annotation, trajectory — without managing pipelines.
The Workflow
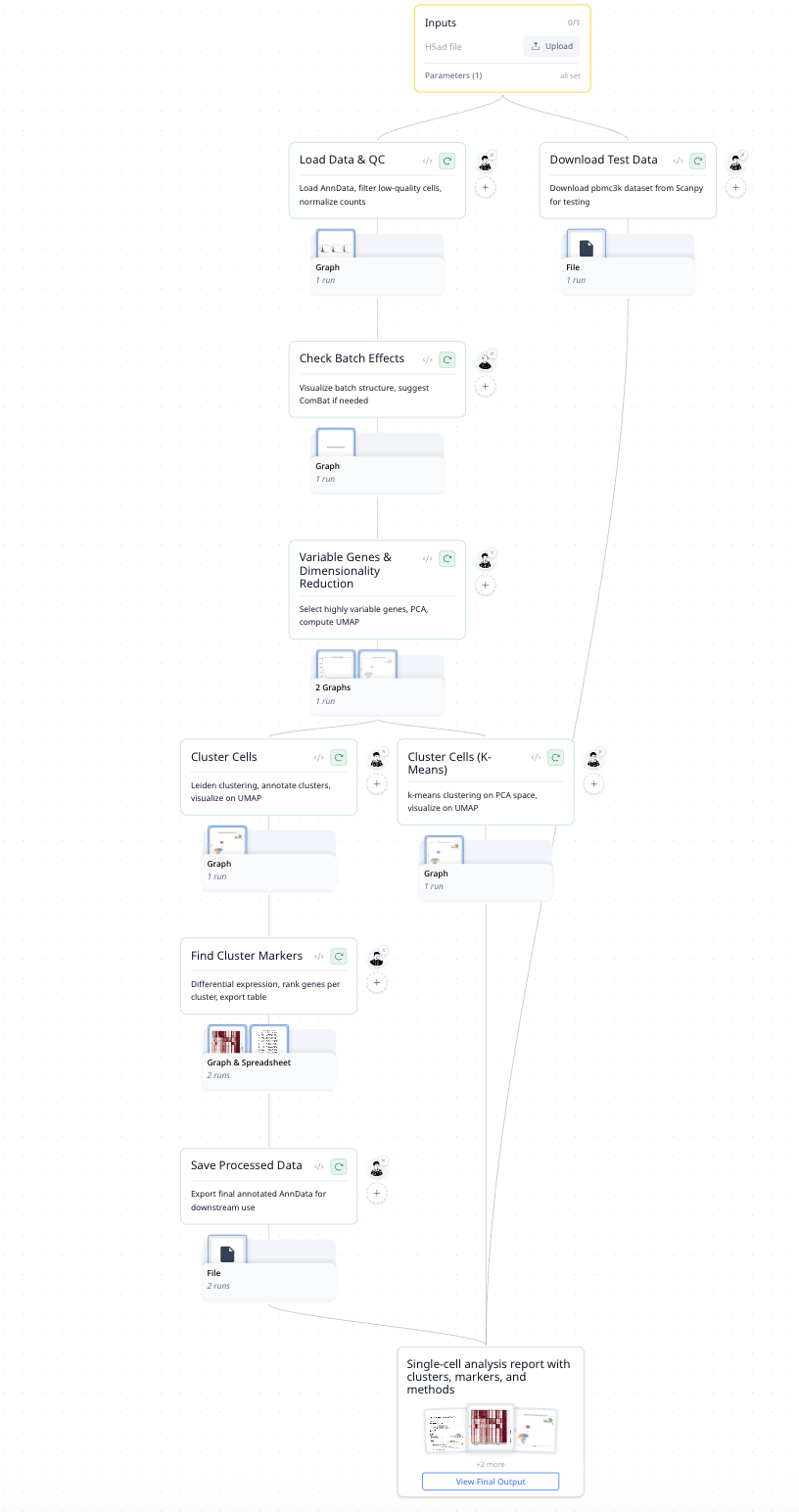
Each step produces a figure — QC violin plots, UMAP embeddings, marker heatmaps — so you always see what happened and why.
| Step | What It Does |
|---|---|
| Data Ingestion | Loads count matrices and cell metadata |
| Quality Control | Filters cells by mitochondrial content, gene counts, doublet detection |
| Normalization | Log-normalization and highly variable gene selection |
| Batch Correction | Applied only when batch effects are detected — skipped if unnecessary |
| Dimensionality Reduction | PCA followed by UMAP embedding |
| Clustering | Leiden community detection with tunable resolution |
| Cell Type Annotation | Marker-based annotation with literature citations |
| Trajectory Analysis | Pseudotime ordering to trace cell state transitions |
How It Was Built
"Build a single-cell RNA-seq analysis copilot. Walks through QC, clustering, annotation, differential expression. Use Scanpy. Validate on PBMC 3k as a test case. Every step should produce a figure."
MorphMind created the full 8-step pipeline and validated it end-to-end on a real dataset before the user ran their own data.
Why This Works Better Than a Chatbot
Ask a chatbot to "analyze my single-cell data" and you get a Scanpy script. Copy-paste it, run it, debug it — and if the clustering looks wrong, start over. That's the problem:
- Batch correction ran when it shouldn't have — but you can't tell because the chatbot gave you one monolithic script. Was it the normalization? The correction? The resolution parameter? You'd have to add print statements and re-run everything.
- Cell type annotations look off — but the QC and clustering were fine. In a pipeline chatbot, you re-run the whole thing. Here, you re-run just the annotation step with different markers.
- You told it "always use Leiden over Louvain" last week — and today it used Louvain again. Chatbots don't carry your preferences. A workflow agent remembers: your preferred algorithms, your quality thresholds, your lab's naming conventions.
| The Problem | Workflow Approach |
|---|---|
| One script — if clustering is wrong, re-run everything | Each step independent — re-run just clustering |
| No way to tell where the error is | Every step produces a figure for inspection |
| Forgets your preferred parameters every session | Rules persist: "if silhouette < 0.3, re-adjust resolution" |
| Batch correction always runs | Conditional: only when detected and biologically inappropriate |
Example Prompts
Upload my 10X count matrix. What cell types are present?
Are there batch effects across my donors? If so, correct them and re-cluster.
Show me the top marker genes for each cluster and annotate the cell types.
Run trajectory analysis — how do cells transition from progenitor to mature state?
Frequently Asked Questions
Can AI do single-cell RNA-seq analysis?
Yes. This agent runs a full Scanpy-based pipeline — QC, normalization, clustering, annotation, and trajectory — with each step producing visual output. It handles real datasets like PBMC 3k out of the box.
Is AI-generated cell type annotation reliable?
The agent uses marker-based annotation with literature citations — each label links to supporting evidence. A separate validation check cross-references annotations against known cell type databases to flag uncertain calls.
What is an automated scRNA-seq pipeline?
A system that runs the standard single-cell analysis workflow (filtering, normalization, clustering, annotation) without requiring the user to write or debug code. This agent adds steerability: you can adjust any step's parameters through conversation and the changes persist.